ORCA中一些性质计算
Properties
单点能(Single point energies)
单点能是薛定谔方程的最低能量解。
有不同的方法计算单点能,同时有不同的计算精度。
Hartree-Fock(HF)
最早开发的近似计算之一,不包括所谓的动态或者静态相关性。
例:
1 | !HF DEF2-SVP |
DEF2-SVP为基组
HF是最老和最简单的方法,他是CCSD、CASSCF方法的基础
Unrestricted HF(UHF)
如果自旋多重度超过1,对于三元组则为3,可以执行UHF计算
在计算中,必须检查结果的自旋污染
如果 S2(写为 <S**2>)算子的期望值与理想值显着不同,则可能是您的系统只能使用多参考计算(例如 CASSCF)来处理
1 | !UHF DEF2-SVP |
1 | ## 结果 |
SCF加速(RIJDX和RIJCOSX)
使用`RI`选项计算库伦积分,从而加速求解SCF方程(计算能量方程)
在此情况下,还需要额外的辅助基组设置。
对于DEF2基组家族,默认设置为`DEF2/J`
1
!HF DEF2-SVP DEF2/J RIJDX
如果选择的基组没有相应的辅助基组,可以使用`AUTOAUX`标志设置自动生成辅助基组
1
!HF 6-31+G(d,p) AUTOAUX RIJDX
为了更快的加速,COSX算法可以和RI一起使用,从而有效的计算交换积分。
密度泛函理论(DFT)
通过从可用的函数中选择任何函数计算,获得DFT能量
1
2
3
4
5
6
!B3LYP DEF2-SVP
* xyz 0 1
O -3.56626 1.77639 0.00000
H -2.59626 1.77639 0.00000
H -3.88959 1.36040 -0.81444
*
注:注意检查自旋污染,DFT结果可能会因为密度泛函的选择而异,在使用之前应进行检查,切勿比较不同方法的能量。
RIJCOSX仅在使用混合密度泛函时才有意义,同时还计算了HF交换
> RIJDX近似是非混合密度泛函的默认值
>
> RIJCOSX是混合密度泛函的默认值。
>
> 如不使用,可以使用`!NORI`或`!NOCOSX`
MP2微扰理论
MP2是一种后HF算法,以HF为基础,并包含丢失的动态相关性。
使用RI近似或者DLPNO算法
1
2
!RI-MP2 cc-pVTZ cc-pVTZ/C
!DLPNO-MP2 cc-pVTZ cc-pVTZ/C
因为是相关算法,对于`RI`部分需要一个特殊的基组`/C`,对于所有的基组来说不易得到,但是可以使用`AUTOAUX`获得
1
2
3
4
5
6
!DLPNO-MP2 6-311++G(2d,2p) AUTOAUX
* xyz 0 1
O -3.56626 1.77639 0.00000
H -2.59626 1.77639 0.00000
H -3.88959 1.36040 -0.81444
*
Coupled Cluster (CC)耦合簇
耦合簇计算,特别是CCSD(T)变体,是单点能的黄金标准。
在常用的公式中,从计算角度看他们的成本非常高
ORCA拥有DLPNO变体,该变体效率更高,对于大型体系有着几乎线性尺度的增长。
为了使用DLPNO,只需在输入中设置DLPNO-CCSD(T)并选择合适的基组(同时需要设置/C基组或者AUTOAUX)
1
2
3
4
5
6
!DLPNO-CCSD(T) cc-pVTZ cc-pVTZ/C
* xyz 0 1
O -3.56626 1.77639 0.00000
H -2.59626 1.77639 0.00000
H -3.88959 1.36040 -0.81444
*
注:检查输出值`T1 diagnostic value`
从经验法则来说,对于`T1 diagnostic value`大于0.02,结果不可信,HF可信度变低
在ORCA中有很多不同的耦合簇和耦合对方法
`RIJCOSX`算法可被用于加速MP2和CC计算
几何优化(Geometry optimization)
基础用法
优化结构:通过给定的算法寻找拥有最小总能量的几何构型
第一步:使用软件给定初始构型,如:Avogadro画出结构并使用简单的优化得到可信的原子排布
第二步:使用Avogadro设置ORCA输入,保存文件为.xyz格式,或者直接在输入文件中写入坐标
如果想使用DFT优化构型,选择方法和基组并添加OPT标志。
OPT命令为几何优化命令
1 | !B3LYP DEF2-SVP OPT |
如果设定正确,可以在输出中看到优化参数:
1 | ***************************** |
然后运行SCF,梯度运算,并给出当前步骤的相关信息
1 | .--------------------. |
当猜测的几何形状很差时,第一步没有达到收敛标准,会根据默认算法(拟牛顿法)修改几何构型
1 | --------------------------------------------------------------------------- |
几何构型的修改会不断迭代更新,知道满足标准
1 | .--------------------. |

尽管最初的猜测似乎不错,但正如人们所见,B3LYP 函数预测的几何形状与给定的输入有些不同。最初的猜测是胺基团处于更平面的构型中,该构型通过优化以及键长的修正而固定
在几何优化之后,会输出一个basename.xyz文件,其中包含最终几何坐标,用于后续计算
你还可以看到分子的能量如何随着优化而进行下降,最后趋于收敛
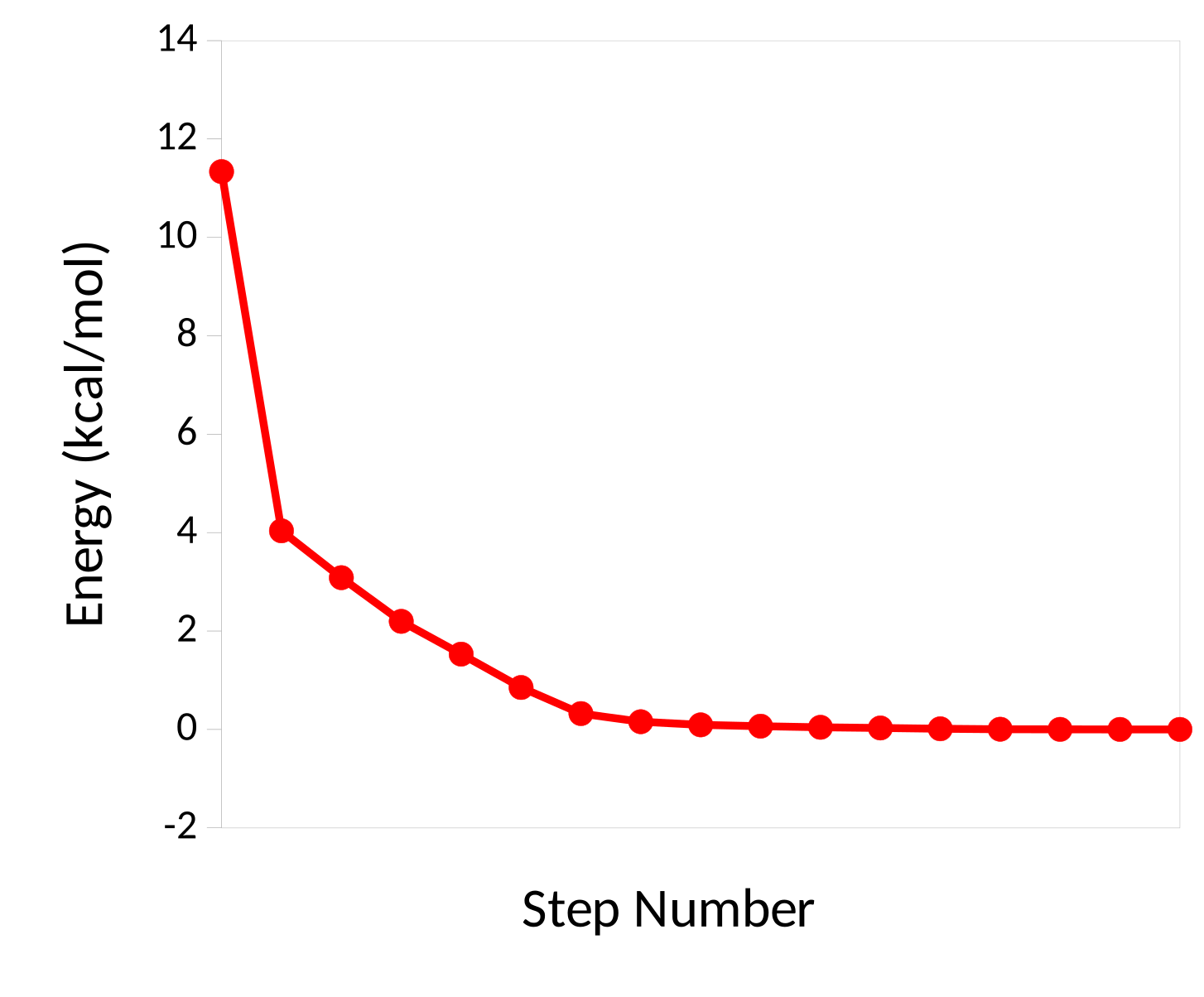
如何判断达到真正的最小值
鞍点的梯度范数也为零,或者梯度可能很低,但还是不够低
可以通过对计算出的最后几何构型计算振动频率来检查是否合理。在主要输入中添加关键词 FREQ
1 | !B3LYP DEF2-SVP OPT RIJCOSX FREQ |
在计算出准确的Hessian矩阵后,将打印频率
1 | ----------------------- |
除了第一次平移和旋转外的所有频率都是正的,这意味着我们确实有一个真正的最小值。
所有正频率都保证局部拥有最小值,但并不是全局最小值
数值梯度
最终生成的几何构型质量取决于你选择的方法。如果你想为当前没有解析梯度的方法优化几何结构你可以使用NUMGRAD标志使用数值梯度计算。
使用数值梯度,耗时会较长
1 | !DLPNO-CCSD(T) cc-pVTZ cc-pVTZ/C OPT NUMGRAD |
如何去除负频率
在几何优化运行后,通常会出现一个或多个负频率,特别是如果你的系统具有弱分子间相互作用,可能计算卡在了鞍点上,或者优化标准不够好
如果系统拥有很多小的负频率,你需要设置更为紧密的约束!TIGHTOPT,,在更严重的情况下,使用!VERYTIGHTOPT
如果您的系统有一个大的负频率,它可能比最小值更接近鞍点。最好的解决方案是采用您的最终结构并将其移向该模式的方向。
DET和OPT讨论
DFT 在分子间或弱相互作用中表现不佳。建议使用 D3 或电荷相关的 D4修正:
1 | !B3LYP DEF2-SVP OPT D3 |
设定将校正能量和梯度,输出更好的结果
振动频率(Vibrational frequencies)
在优化构型后,你将计算系统的振动频率并绘制振动动画。
使用醋酸为例,使用DFT B3LYP:
1 | !B3LYP DEF2-SVP OPT FREQ |
也可以在优化后立即运行
1 | !B3LYP DEF2-SVP OPT FREQ |
在几何优化成功之后,进行频率计算。当Hessian矩阵计算开始时,会输出以下信息:
1 | ------------------------------------------------------------------------------- |
然后计算所有必要的术语,完成后,可以直接看到以cm-1为单位的振动频率
1 | ----------------------- |
频率前几行均为0,他们对应着旋转和平移模式。线性分子应为5个0,非线性分子为6个0,剩余的对应实际振动模式。
缩放频率
如果你想在计算热力学函数之后所有频率乘以某个数字,可以使用
1 | %FREQ SCALFREQ 1.035 END |
所有的频率都将乘以1.035
时刻检查振动频率。如果几何构型最小,应当没有负频,如果是过渡态,有且仅有一个负频
可视化
振动模式可以使用Avogadro查看,打开.out文件,在右侧可以看到一个面板
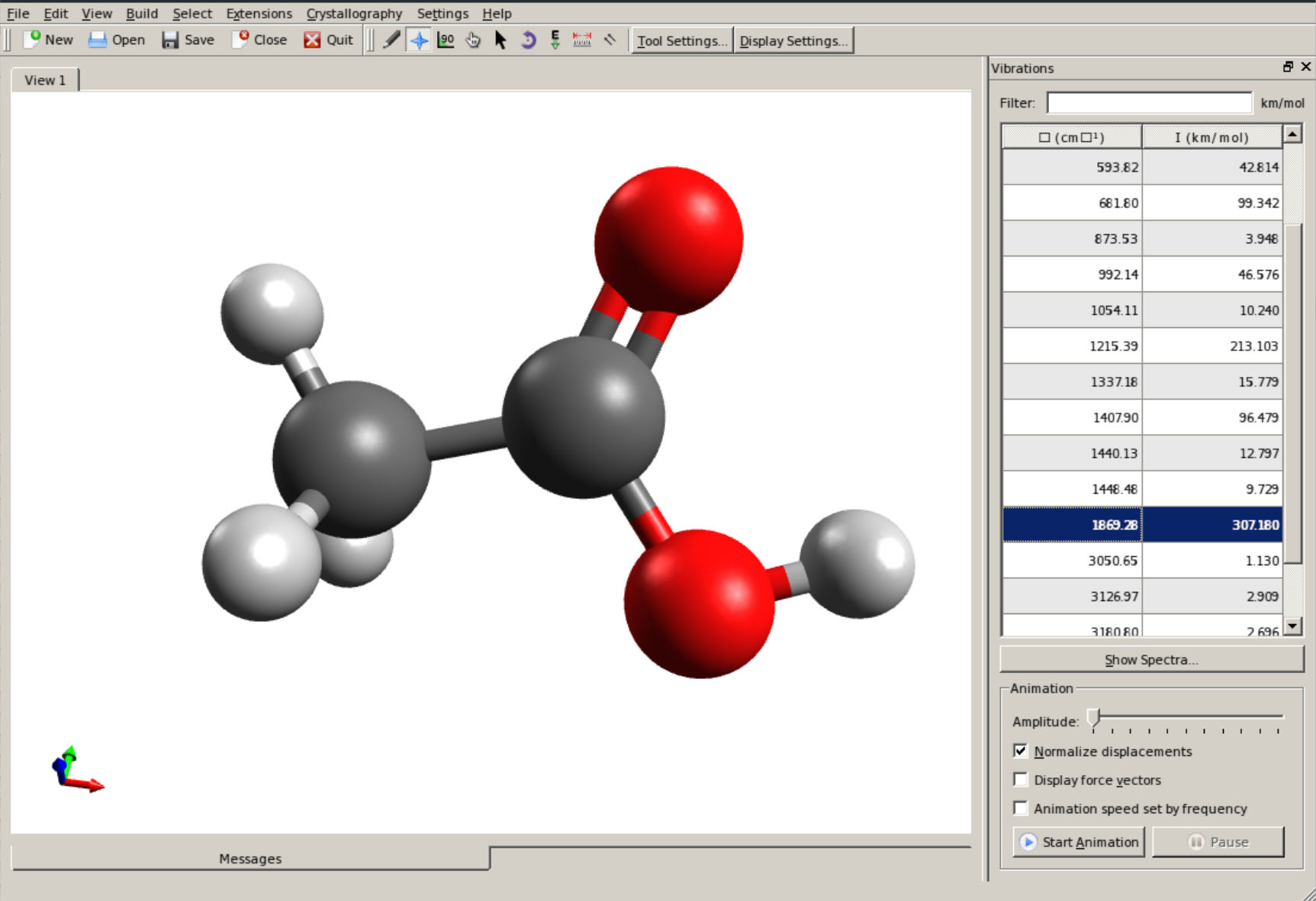
你可以选择一个振动频率并单击Animate查看与该频率对应的动画。如果点击约~1800cm-1, 这是有机化学中C=O键拉伸模式,
~3150cm-1对应C-H键拉伸
消除负频
在计算之后,如果有一个或多个负频,意味着结构不稳定
以下时会产生负频的输入文件:
1 | !B3LYP DEF2-SVP OPT FREQ |
优化后可以得到输出:
1 | ----------------------- |
使用所谓的虚模式,这意味着几何构型会收敛到鞍点,并且沿着该模式的方向移动会降低分子的能量。解决问题的方法时打开Avogadro,点击-85.0cm-1负频。
现在,如果您在分子位移时单击“暂停”,您将获得远离鞍点的几何形状。然后可以保存这些坐标并重新开始计算,最终收敛到最小值:
1 | !B3LYP DEF2-SVP OPT FREQ |
数值频率
ORCA中并不是所有方法都可以计算Hessian矩阵,但是都可以用numerical Hessian计算。使用NUMFREQ
1 | !RI-B2PLYP DEF2-SVP DEF2-SVP/C NUMFREQ |
在 6N 次位移后,Hessian 将通过中心差异方法计算,其中 N是您系统中的原子数。这可以完全并行化,有时实际上是比分析 FREQ 计算更好的选择,具体取决于您的内存。
重启NUMFREQ计算
如果系统较大或者计算较重,计算可能需要很长时间,如果终断,可以通过以下方法计算
1 | !RI-B2PLYP DEF2-SVP DEF2-SVP/C NUMFREQ |
如果basename.hess文件与输入文件存放在同一文件夹,计算将从停止地方重新开始。
热力学(Thermodynamics)
在成功计算振动频率后,你可以进行热力学函数计算,例如焓(enthalpy H),熵(entropy S),吉布斯自由能(Gibbs free energy G)
对一些丁二烯异构体计算热力学函数
1 | !B3LYP DEF2-TZVP OPT FREQ |
1 | !B3LYP DEF2-TZVP OPT FREQ |
热力学功能
假设输入文件写入 !FREQ,在频率和正常模式之后会立刻看到计算热力学部分
这些属性将在给定的温度和压力下计算,假设气相分子并使用统计力学。
1 | !B3LYP DEF2-TZVP OPT FREQ |
热力学开始
1 | -------------------------- |
内能(inner energy U)
1 | ------------ |
Electronic energy 是从最终单点能量得到的
Zero point Energy 零点能量
thermal correction 热力学校正
rotational 旋转
vibrational 振动
translational degrees of freedom 平移自由度
总校正能量是用于将Eel转换为U时添加进最终单点能的能量
焓(Enthalpy H)= U + kBT
热焓校正
1 | -------- |
熵(Entropy S)
1 | ------- |
吉布斯自由能(Gibbs free energy G)
1 | ------------------- |
G-E(el)是在特定温度压力下,添加进电子能量项用于将Eel转换为G0
G-E(el)仅取决于几何结构和频率,能用低等级运算例如DFT,然后结合更高等级的方法计算Eel,例如DLPNO-CCSD(T)获得更好的预测结果
实验比较
DFT直接计算吉布斯自由能
1 | !B3LYP DEF2-TZVP OPT FREQ |
DLPNO-CCSD(T)计算Eel
1 | !DLPNO-CCSD(T) aug-cc-pVTZ AUTOAUX RIJCOSX |
将DFT算出的G-E(el)添加到更高级的算法结果中,形成更好的吉布斯自由能
不同温度的热力学
计算不同温度下的热力学性质
1 | %FREQ TEMP 77, 298, 330, 450 END |
热力学函数被不同的温度标题区分
1 | -------------------------- |
在计算结束之后改变温度
如果在计算结束之后,还需进一步计算热力学函数,可以设置如下输入:
1 | !PRINTTHERMOCHEM |
可以从重新计算中节省大量时间
结构
gem-butadiene
2
3
4
5
6
7
8
9
10
11
12
C -0.17392 0.62436 -0.39435
C 0.14450 -0.70205 -1.02166
C 1.00254 1.50776 -0.09468
H 0.65348 -0.55631 -1.97995
H -0.75651 -1.29570 -1.20886
H 0.79819 -1.28546 -0.36521
H -2.27870 0.35250 -0.33839
H -1.65494 1.95886 0.33562
H 1.55177 1.73378 -1.01439
H 0.69898 2.45896 0.35516
H 1.68283 1.00994 0.60389trans-butadiene
2
3
4
5
6
7
8
9
10
11
12
C -0.04707 -0.10202 0.22754
H 1.76922 0.66709 1.03514
H 1.25468 1.48282 -0.45237
H 1.94420 -0.14853 -0.53097
C -1.18840 0.46276 -0.19477
C -2.53838 -0.15441 -0.02414
H -3.00427 -0.30734 -1.00238
H -3.17998 0.50979 0.56286
H -2.49026 -1.12154 0.48617
H -0.07366 -1.06762 0.72803
H -1.16180 1.42834 -0.69529cis-butadiene
2
3
4
5
6
7
8
9
10
11
12
C 0.03276 -0.80512 0.28503
H 1.68200 0.56889 -0.06324
H 1.92979 -1.08298 -0.63914
H 1.99954 -0.72748 1.09552
C -1.01110 0.03009 0.16420
H -0.18277 -1.85160 0.49931
C -0.98494 1.49624 -0.12334
H -2.00868 -0.39072 0.28765
H -1.47672 2.03930 0.68986
H 0.02271 1.90131 -0.23449
H -1.53322 1.69981 -1.04849
隐式解模型(inplicit solvation models)
隐式溶剂模型:在计算中包含溶剂效应的一种方法
主要讨论ORCA中两个主要的选项
模型解释:溶质被放置在一个大致分子形状的空腔中。溶剂由与空腔表面上电荷相互作用的连续体描述,而连续体又由溶质决定。
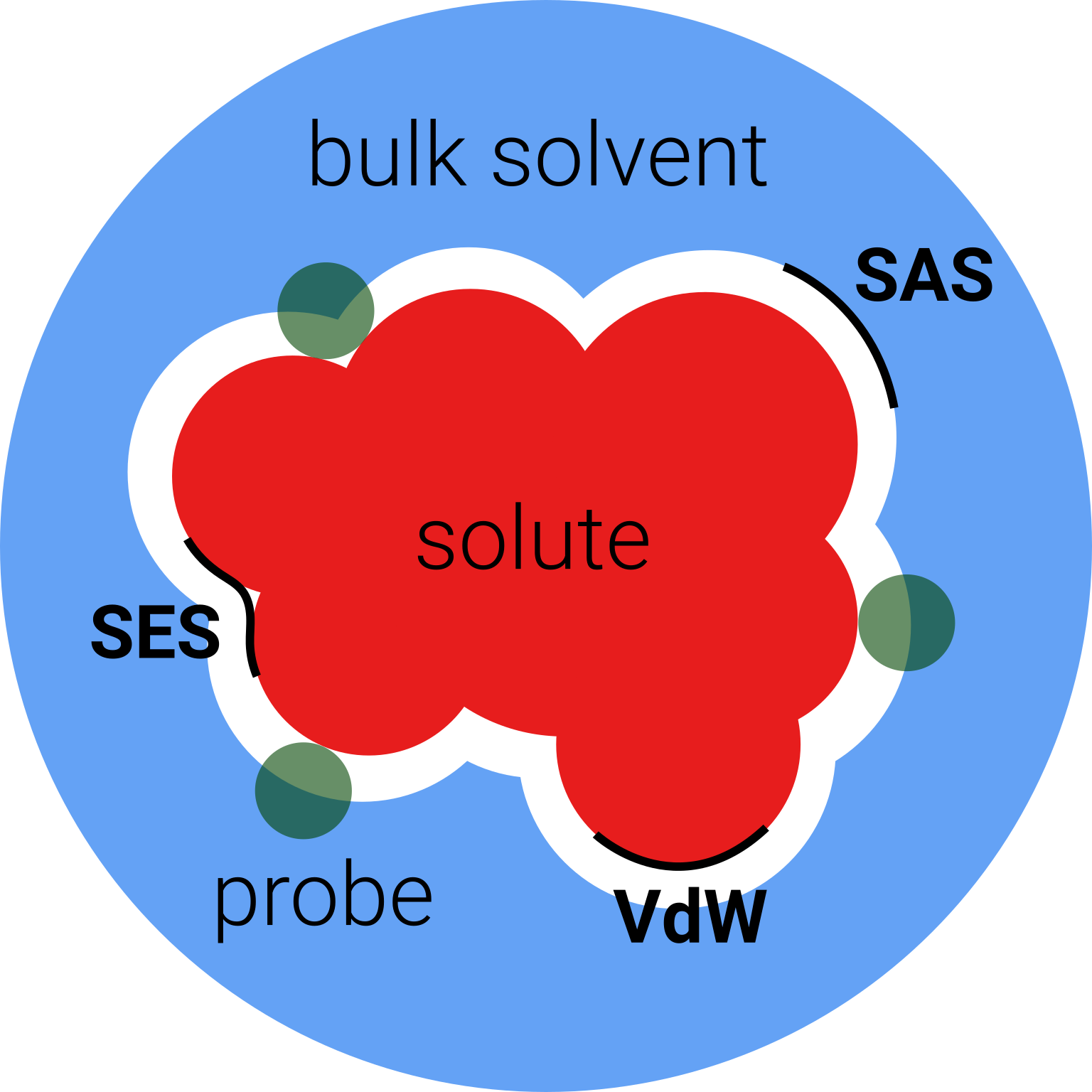
probe是理想的溶剂分子
分子空腔被定义为
- Van der Waals surface(VDW),从vdw半径构建
- Solvent-Accessible Surface(SAS),溶剂可及表面,由probe中心定义
- Solvent-Excluded Surface(SES),无溶剂表面,沿着在 vdW 表面滚动的溶剂探针球体的内部部分获得
溶剂能由两部分组成,静电势electrostatic(ΔGENP)和腔色散cavitydispersion(ΔGCDS)
ΔG0solv=ΔGENP+ΔGCDS
Conductor-like Polarizable Continuum Model (CPCM)
在 CPCM 方法 中,本体溶剂被视为类似导体的可极化连续体,定义该方法的主要参数是介质的__折射率__和__介电常数__
由介质和分子表面电荷相互作用产生的静电贡献 (ΔGENP) 包含在 SCF 计算中 - 这样您甚至可以获得“溶剂化”轨道 - 并且腔项 (ΔGCDS) 可以从更复杂的方案中获得。
ORCA 有一个预定义溶剂列表,可以使用以下命令调用这些溶剂:!CPCM(solvent)
| Solvent Name | Dielec. Const.介电常数 | Refrac. Index折射率 |
|---|---|---|
| Water | 80.4 | 1.33 |
| Acetone | 20.7 | 1.359 |
| Acetonitrile | 36.6 | 1.344 |
| Ammonia | 22.4 | 1.33 |
| Benzene | 2.28 | 1.501 |
| CCl4 | 2.24 | 1.466 |
| CH2Cl2 | 9.08 | 1.424 |
| Chloroform | 4.9 | 1.45 |
| Cyclohexane | 2.02 | 1.425 |
| DMF | 38.3 | 1.430 |
| DMSO | 47.2 | 1.479 |
| Ethanol | 24.3 | 1.361 |
| Hexane | 1.89 | 1.375 |
| Methanol | 32.63 | 1.329 |
| Octanol | 10.3 | 1.421 |
| Pyridine | 12.5 | 1.510 |
| THF | 7.25 | 1.407 |
| Toluene | 2.4 | 1.497 |
如果想包括其他溶剂,可以输入下列参数:
1 | %CPCM EPSILON 80.4 |
例:使用单点能计算水中阿司匹林分子
1 | !BP86 DEF2-SVP CPCM(WATER) |
1 | # 阿司匹林 |
CPCM可以使用解析梯度以及Hessian矩阵,所以CPCM可以和OPT以及FREQ一起使用
在SCF之前,打印一个包含所有CPCM相关信息的标题
1 | -------------------- |
在SCF计算结束之后,会有CPCM相关信息的汇总
1 | Components: |
ORCA默认不会为CPCM计算或添加空腔
将DFT和后HF结合
HF密度质量低于从DFT获得的质量,因此使用HF获得的CPCM校正也是如此
通常做法:从某些计算(如 DFT)中获取 ΔGENP(CPCM 电介质)和 ΔGCDS(自由能(cav+disp)),并将其添加到使用该方法获得的总能量中,例如如果您想将 CPCM 与 DLPNO-CSSD 结合使用。
Gaussian point charges
通常点电荷会导致能量不稳定,例如两点太近而崩溃。在ORCA,使用gaussian电荷来获得一个平滑的势能面。
在ORCA5.0中,默认设置为
1 | %CPCM SURFACETYPE VDW_GAUSSIAN END |
但是SES表面也可以被设置为:
1 | %CPCM SURFACETYPE GEPOL_SES_GAUSSIAN END |
Universal Solvation Model (SMD)全溶剂模型
SMD方法可被认为是CPCM的改进版
SMD使用完整的溶质电子密度来计算腔色散贡献,而不仅仅是区域
SMD需要更多的参数,这使得SMD对未知溶剂的灵活性兼容性较差,但是目前ORCA拥有179种溶剂可用。
使用方法:
1 | %CPCM SMD TRUE |
例:
1 | !BP86 DEF2-SVP |
SMD最开始输出与CPCM相似
1 | SMD-CDS solvent descriptors: |
可以使用ΔGCDS (Gcds) 项和ΔGENP (CPCM Dielectric) 校正进一步计算的能量。
Example-辛醇/水分配系数
使用SMD模型,预测药物Diazepam在octanol/water中的分配系数
$$
P_{o/w}=\frac {[solute]{octanol}} { [solute]{water}}
$$
P可以从平衡常数以及溶剂在辛醇和水中的吉布斯自由能差异关系中得到 ΔGoo/w=ΔGoo−ΔGow
$$
logP_{o/w}=\frac{-\Delta G^o_{o/w}}{2.303RT}
$$
首先,优化在不同溶剂中的几何构型
1 | !B3LYP DEF2-SVP OPT NUMFREQ D4 |
得到每个溶剂化化合物的吉布斯自由能及其几何形状
经过计算,得到3.41kcalmol-1的能量差,对应logP=2.5,实验值为2.99.
气液相转变
$$
\Delta G^o_{solv}=\Delta G {ENP}+\Delta G{CDS}+\Delta G^o_{conc}
$$
在1atm 298k时,从1molL-1气相转变为液相(需要计算G0,默认使用FREQ)
$$
\Delta G^o_{conc}=RTln(24.5)=1.89kcalmol^{-1}
$$
在预测溶液热力学时,不要忘记加G0.
CPCM 和COSMO
ORCA没有COSMO接口,但是可以使用CPCMC关键词来调用COSMO epsilon
1 | !B3LYP DEF2-SVP CPCMC(WATER) |
Correlation Energy
Correlation Energy被定义为高等级理论方法(MP2,CCSD)与参考HF方法之间的能量差
他是精确总能量的重要指标,对于预测特性或能量差有重要意义
在ORCA中,有几种方法来计算或者考虑Correlation Energy。
在此讨论三种
- Møller–Plesset perturbation theory (MP2);
- double-hybrid DFT (DHDF)
- coupled cluster (CC), using the singlet-triplet gap of methylene as a benchmark
讨论的范例,三重态比单重态更加稳定,HF方法误差极大,并且实验特性测量的非常准确
Møller–Plesset perturbation theory (MP2)
使用DHDF B2PLYP计算单重态和三重态的参考几何,能够准确的体现实验得到的几何坐标
1 | !RI-B2PLYP DEF2-TZVPP DEF2-TZVPP/C OPT |
计算得出的三重态键角为134°,与实验相符
接着,使用MP2和先前针对单重态和三重态优化的结构计算绝热单重态-三重态之间的能量差
1 | # 单重态 |
1 | # 三重态 |
相关方法的准确性很大程度上取决于基组的大小,原则上选取更大的基组作为目标。
SCF之后将显示MP2header
1 | ------------------------------------------------------------------------------ |
积分详细信息
1 | ------------------------------- |
最后输出单重态相关能量
1 | ----------------------------------------------- |
生成最终能量
1 | ------------------------- -------------------- |
单重态减去三重态能量得到 ΔEMP2=14.24kcal/mol,比实验值高5
ΔEHF=28.25kcal/mol
Spin-component-scaled MP2 (SCS-MP2)
自旋分量级MP2
另一个版本的MP2理论为SCS-MP2,总体上由于MP2,可以如下调用
1 | !SCS-MP2 DEF2-QZVPP |
ΔESCS−MP2=7.92kcal/mol,比MP2更好
使用 resolution of identity(RI)
MP2或SCS-Mp2计算可以使用库伦积分的恒等近似分辨率来加速
1 | !RI-MP2 DEF2-QZVPP DEF2-QZVPP/C |
此例中,相关方法需要使用特殊的辅助基/C,可以随时使用AUTOAUX自动生成
任何近似值都存在固有误差,但误差相对较小。单重态RI-MP2最终能量为:
1 | ------------------------- -------------------- |
RIJDX 和 RIJCOSX 近似也可用于 SCF 部分,通过在主输入中使用 !RIJDX 或 !RIJCOSX 进一步加速计算。
DLPNO scheme
Domain-based Local Pair Natural Orbital (DLPNO): ORCA团队开发了相关方法的特殊近似
相关能量可以用电子对能量计算,并且这些能量只能在每对的某个域内有效计算,使计算更加高效,并恢复了大约99%的相关能量。
1 | !DLPNO-MP2 DEF2-QZVPP DEF2-QZVPP/C |
输出有额外的输出信息,与轨道定位有关
1 | ------------------------------------------------------------------------------ |
MP2运算
1 | ---------------------------- |
相关能量输出,与常规MP2 99.99%相似
1 | ------------------------------------------------------ |
Calculations using DLPNO also always require a /C basis.
Double-hybrid density functionals (DHDF) 双混合密度泛涵
DFT already has a component of correlation energy to them, added by the exchange-correlation functional, but it was shown that by adding a certain amount of the MP2 correlation energy the results can be improved even further
这些具有MP2组件的泛函成为双杂化泛函,他们同时包括HF交换和MP2相互关系,他们可以被调用,同时使用RI- DLPNO-近似
1 | !B2PLYP DEF2-QZVPP |
The output follows a regular DFT calculation, with an MP2 calculation being done after the SCF. Finally all energies are added according the functional definition and the final energy is printed. For RI-B2PLYP, the singlet-triplet gap for methylene is ΔEB2PLYP=11.73kcal/mol, better than the simple MP2.
Double-hybrid spin-component-scaled density functionals (DSD-DF)
Following the same logic used for SCS-MP2, the different spin components of the MP2 energy can be scaled for the DHDF, which usually results in even better predictions. These functionals have DSD- in front of their name, for instance:
1 | !DSD-PBEB95 DEF2-QZVPP |
所有的方法均采用解析梯度,对于几何优化十分有用
Coupled cluster methods (CC)
耦合聚类方法
量子力学中获得相关能量的最准确方法之一为Coupled cluster methods(CC),特别是单参考问题中能准确计算能量的CCSD(T)方法
然而,尺度会随着电子数量的增加而增加,并且很快会变得不现实,其应用范围限制在20个原子左右
在ORCA中,使用DLPNO方案降低尺度问题,现在可以计算具有数百个原子的真实大小的分子
1 | !DLPNO-CCSD DEF2-QZVPP DEF2-QZVPP/C |
after the regular HF output, the Matrix Driven CI module starts (MDCI)
1 | ------------------------------------------------------------------------------- |
接下来输出位置、筛选、积分变换,CCSD计算
1 | ------------------------------------------------ |
当收敛之后,汇总输出不同的能量组成以及诊断信息
1 | ---------------------- |
注意检查T1 diagnostic信息,T1诊断信息值大于0.02,结果不可信,并且HF参考会变差。
The predicted energy difference here is ΔECCSD=10.54kcal/mol,better than the MP2, but still with some error. If one runs the same calculation using the even more accurate DLPNO-CCSD(T) instead, that difference goes down to ΔECCSD(T)=9.70kcal/mol, which is almost exactly like the experimental value!
Extrapolation to the CBS limit 外推到CBS限制
As mentioned before in the MP2 section, these methods require large basis set to be really accurate, and their true power show up only when close to the basis set (CBS) limit. Even so, one does not need to use an immense basis to get there, it is also possible to extrapolate the CBS from lower basis set calculations
之前在 MP2 部分中提到的,这些方法需要大的基组才能真正准确,并且只有在接近基组 (CBS) 限制时才会显示它们的真实功效。即便如此,也不需要使用巨大的基来达到目标,也可以从较低的基组计算中推断出 CBS
计算接近CBS的DLPNO-CCSD(T)能量
1 | !DLPNO-CCSD(T) EXTRAPOLATE(3) cc-pVQZ/C |
默认不自动/C基组,选择大的基组使合适的。
Going back to methylene triplet-singlet gap, the extrapolated energy difference is ΔECCSD(T)−CBS=9.26kcal/mol, which is actually within the experimental error.
总结计算结果
| Method | ΔETS | Error |
|---|---|---|
| HF | 28.25 | 19.13 |
| B3LYP | 11.74 | 2.62 |
| MP2 | 14.24 | 5.11 |
| SCS-MP2 | 7.92 | -1.22 |
| RI-B2PLYP | 11.73 | 2.60 |
| RI-DSD-PBEB95 | 11.63 | 2.50 |
| DLPNO-CCSD | 10.54 | 1.42 |
| DLPNO-CCSD(T) | 9.70 | 0.58 |
| DLPNO-CCSD(T)-CBS | 9.26 | 0.14 |
| Experiment | 9.12 | ± 0.2 |
Relativistic corrections 相对论校正
当计算包含重原子系统的性质或者几何构型时,相对论效应有很大影响,并且不能忽略。
两种方法相对论校正,零阶正则近似(Zero-Order Regular Approximation ZORA)和Douglas-Kroll-Hess (DKH) Hamiltonian。
他们有些许不同,并且具有特定的优势,必须仔细考虑结果,使用最广泛的是ZORA和其改进型
Including relativistic corrections
使用输入:
1 | !B3LYP ZORA ZORA-DEF2-TZVP |
Note that here we are using specific basis for each method, named ZORA- or DKH-DEF2-TZVP. This is necessary because these basis have been specifically designed for these all-electrons calculations, and the relativistic correction should NOT be used together with the regular basis or pseudopotentials.
RI and ZORA/DKH
使用RI方法加速SCF,另一个特殊基组/J被使用
1 | !B3LYP ZORA ZORA-DEF2-TZVP RIJDX SARC/J |
For instance, the SARC/J auxiliary basis can be used for all the ZORA or DKH-DEF2 basis. If no specific basis is available, then one can always use AUTOAUX to automatically generate one.
The SARC/J basis were optimized for RI on the SCF part, not the MP2 or higher-level correlated methods! For correlation specific /C basis consult the manual and in case of abscence use !AUTOAUX.
Example : Hg dimer
Let’s test the impact of these effects on the Hg dimer, that has an experimental bond length of 3.69Å. This a rather extreme case of a very heavy element in a homodiatomic molecule, however it highlights the importance and magnitude of these effects.
The geometry can be optimized at a regular Double-hybrid density functionals (DHDF) level using the DEF2-TZVP basis, that makes use of pseudopotentials for the core orbitals that try to simulate relativistic effects or using the all-electron ZORA-DEF2-TZVP basis:
1 | !B2PLYP DEF2-TZVP DEF2-TZVP/C OPT |
The first results in a molecule with a 3.84Å bond, and the relativistic ZORA results in 3.78Å , which is in better agreement with the experimental results!
The auxiliary basis for the RIJ approximation used during the relativistic case here was chosen as the appropriate SARC/J.
Geometry optimizations using relativistic corrections turn on by default a one-center approximation by default, that changes the energy values. Do not compare single point energies from those you obtain from an !OPT run, these numbers are be incompatible.