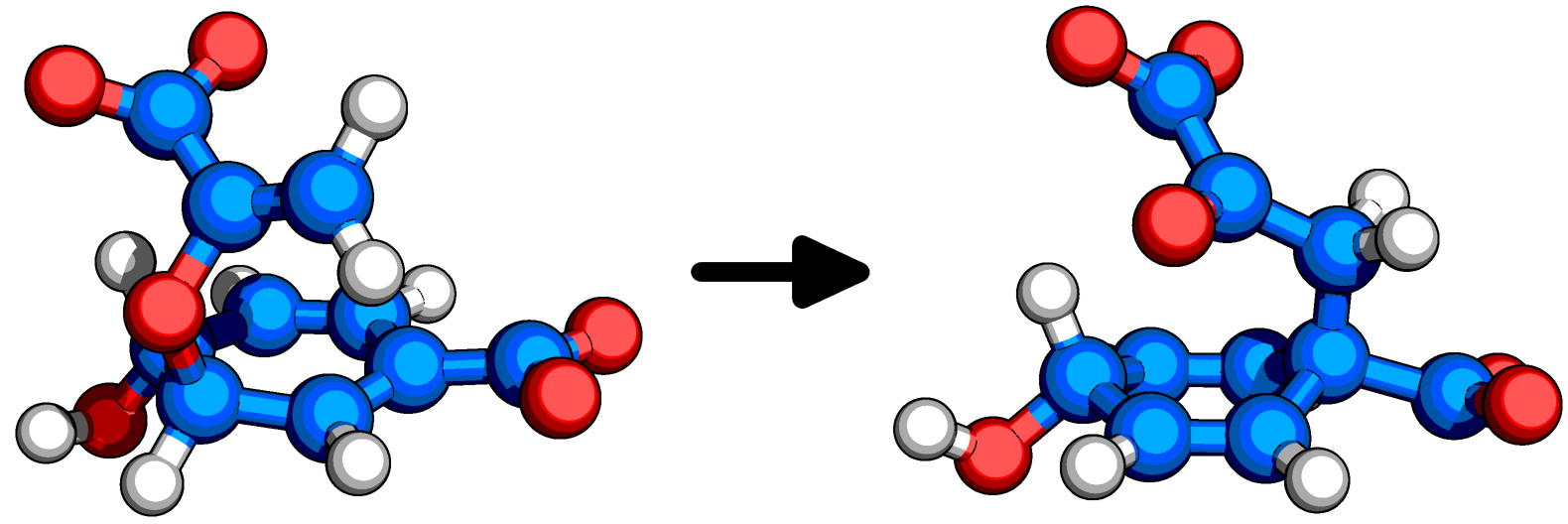
for i in A B C; do antechamber -i Lig${i}.mol2 -o Ligand${i}.mol2 -fi mol2 -fo mol2 -c bcc -pf yes -nc -2 -at gaff2 -j 5 -rn CH${i}; done for i in A B C; do parmchk2 -i Ligand${i}.mol2 -f mol2 -o Ligand${i}.frcmod -s 2; done
# cp2k can be run using a single process: cp2k.sopt em.inp > em.out # or using several processes, where N is the number of processes: mpirun -np N cp2k.popt em.inp > em.out
# em.inp &GLOBAL PROJECT EM PRINT_LEVEL LOW RUN_TYPE GEO_OPT &ENDGLOBAL
&FORCE_EVAL METHOD FIST &MM &FORCEFIELD PARMTYPE AMBER PARM_FILE_NAME complex.prmtop &SPLINE EMAX_SPLINE 1.0E8 RCUT_NB [angstrom] 10 &END SPLINE &END FORCEFIELD &POISSON &EWALD EWALD_TYPE SPME ALPHA .40 GMAX 80 &END EWALD &END POISSON &END MM &SUBSYS &CELL !Setbox dimensions here ABC [angstrom] 84.482435085.205280084.5083700 ALPHA_BETA_GAMMA 909090 &END CELL &TOPOLOGY CONN_FILE_FORMAT AMBER CONN_FILE_NAME complex.prmtop COORD_FILE_FORMAT CRD COORD_FILE_NAME complex.inpcrd &END TOPOLOGY !NA+ isnot recognized by CP2K, so it is necessary to define it here using KIND &KIND NA+ ELEMENT Na &END KIND &KIND NS3 ELEMENT N &END KIND &KIND NS4 ELEMENT N &END KIND &KIND NS1 ELEMENT N &END KIND &KIND NS2 ELEMENT N &END KIND
&END SUBSYS &END FORCE_EVAL
&MOTION &GEO_OPT OPTIMIZER LBFGS MAX_ITER 1000 MAX_DR 1.0E-02 !Convergence criterion for the maximum geometry change between the currentand the last optimizer iteration RMS_DR 5.0E-03 !Convergence criterion for the root mean square (RMS) geometry change between the currentand the last optimizer iteration MAX_FORCE 1.0E-02 !Convergence criterion for the maximum force component of the currentconfiguration RMS_FORCE 5.0E-03 !Convergence criterion for the root mean square (RMS) force of the currentconfiguration
&END &PRINT &TRAJECTORY ! Controls the output of the trajectory FORMAT XYZ ! Formatof the output trajectory is XYZ &EACH ! New trajectory frame will be printed each100 md steps GEO_OPT 200 &ENDEACH &END TRAJECTORY &RESTART ! This section controls the printing ofrestart files &EACH ! A restart file will be printed every 10000 md steps GEO_OPT 500 &ENDEACH &ENDRESTART &RESTART_HISTORY ! This section controls dumping ofuniquerestart files during the run keeping allof them.Most useful if recovery is needed at a later point. &EACH ! A newrestart file will be printed every 10000 md steps GEO_OPT 500 &ENDEACH &END RESTART_HISTORY &END PRINT &END MOTION
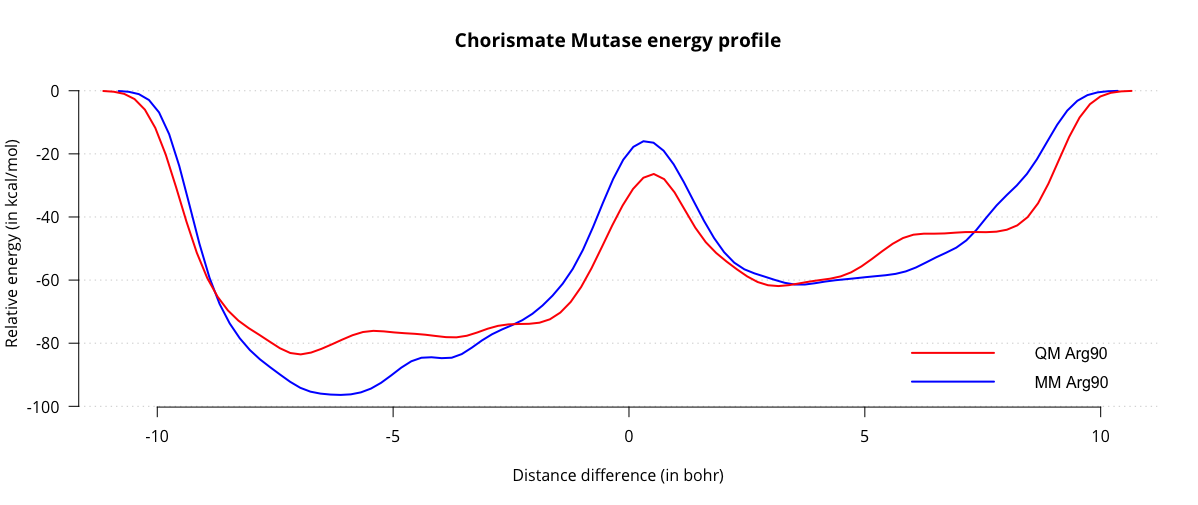
&MOTION &MD ENSEMBLE NVT TIMESTEP [fs] 0.5 STEPS 10000 TEMPERATURE 298 &THERMOSTAT REGION GLOBAL TYPE CSVR &CSVR TIMECON [fs] 10 &END CSVR &PRINT &TEMPERATURE &EACH MD 100 &END EACH &END TEMPERATURE &END PRINT &END THERMOSTAT &END MD &CONSTRAINT &COLLECTIVE COLVAR 1 TARGET [angstrom] 3.55 MOLNAME MOL4 &RESTRAINT K [kcalmol] 10 &END RESTRAINT &END COLLECTIVE &END CONSTRAINT
&PRINT &RESTART_HISTORY ! This section controls dumping of unique restart files during the run keeping all of them.Most useful if recovery is needed at a later point. &EACH ! A new restart file will be printed every 10000 md steps MD 5000 &END &END &RESTART ! This section controls the printing of restart files &EACH ! A restart file will be printed every 10000 md steps MD 5000 &END &END &TRAJECTORY ! Thes section Controls the output of the trajectory FORMAT XYZ ! Format of the output trajectory is XYZ &EACH ! New trajectory frame will be printed each 100 md steps MD 2000 &END &END &END PRINT &END MOTION
&PRINT &RESTART ! This section controls the printing of restart files &EACH ! A restart file will be printed every 10000 md steps MD 25000 &END &END &TRAJECTORY ! Thes section Controls the output of the trajectory FORMAT XYZ ! Format of the output trajectory is XYZ &EACH ! New trajectory frame will be printed each 100 md steps MD 5000 &END &END &RESTART_HISTORY ! This section controls dumping of unique restart files during the run keeping all of them.Most useful if recovery is needed at a later point. &EACH ! A new restart file will be printed every 10000 md steps MD 5000 &END &END &CELL &EACH MD 100 &END &END &END PRINT &END MOTION
&MOTION &MD ENSEMBLE NVT TIMESTEP [fs] 0.5 STEPS 10000 !5000 fs = 5ps TEMPERATURE 298 &THERMOSTAT TYPE NOSE REGION GLOBAL &NOSE TIMECON [fs] 100. &END NOSE &END THERMOSTAT &END MD &FREE_ENERGY METHOD METADYN &METADYN DO_HILLS .FALSE. NT_HILLS 100 WW [kcalmol] 1.5 &METAVAR WIDTH 0.5 !Also known as scale COLVAR 1 &END METAVAR &PRINT &COLVAR COMMON_ITERATION_LEVELS 3 &EACH MD 10 &END &END &END &END METADYN &END FREE_ENERGY
&PRINT &RESTART ! This section controls the printing of restart files &EACH ! A restart file will be printed every 10000 md steps MD 1000 &END &END &RESTART_HISTORY ! This section controls dumping of unique restart files during the run keeping all of them.Most useful if recovery is needed at a later point. &EACH ! A new restart file will be printed every 10000 md steps MD 1000 &END &END &TRAJECTORY ! Thes section Controls the output of the trajectory FORMAT XYZ ! Format of the output trajectory is XYZ &EACH ! New trajectory frame will be printed each 100 md steps MD 1000 &END &END &END PRINT &END MOTION
&MOTION &MD ENSEMBLE NVT TIMESTEP [fs] 0.5 STEPS 60000 !30000 fs = 30 ps TEMPERATURE 298 &THERMOSTAT TYPE NOSE REGION GLOBAL &NOSE TIMECON [fs] 100. &END NOSE &END THERMOSTAT &END MD &FREE_ENERGY METHOD METADYN &METADYN DO_HILLS .TRUE. NT_HILLS 100 WW [kcalmol] 1.5 &METAVAR WIDTH 0.5 !Also known as scale COLVAR 1 &END METAVAR &PRINT &COLVAR COMMON_ITERATION_LEVELS 3 &EACH MD 10 &END &END &END &END METADYN &END FREE_ENERGY
&PRINT &RESTART ! This section controls the printing of restart files &EACH ! A restart file will be printed every 10000 md steps MD 5000 &END &END &RESTART_HISTORY ! This section controls dumping of unique restart files during the run keeping all of them.Most useful if recovery is needed at a later point. &EACH ! A new restart file will be printed every 10000 md steps MD 5000 &END &END &TRAJECTORY ! Thes section Controls the output of the trajectory FORMAT XYZ ! Format of the output trajectory is XYZ &EACH ! New trajectory frame will be printed each 100 md steps MD 1000 &END &END &END PRINT &END MOTION