MMPBSA计算程序开发小记
前言
做任何事,都要知其然,知其所以然。之前使用工具的时候,直接就是一顿莽,根本不知道其底层的逻辑和原理,导致计算频频出错。恰巧使用李老师开发的MMPBSA工具[1]一段时间了,借着这个契机尝试着将此脚本用python复现出来,本文会尽可能地将计算mmpbsa的理论基础,注意事项等讲解清楚。文中可能出现错误,欢迎指正。
本文介绍了使用python计算蛋白-配体结合能的脚本,所需文件(tpr、xtc、ndx)均为gromacs格式。
需要的python库及程序:
- numpy
- pandas
- matplotlib
- prettytable
- multiprocessing
- mdanalysis
- apbs
- gmx
理论与开发
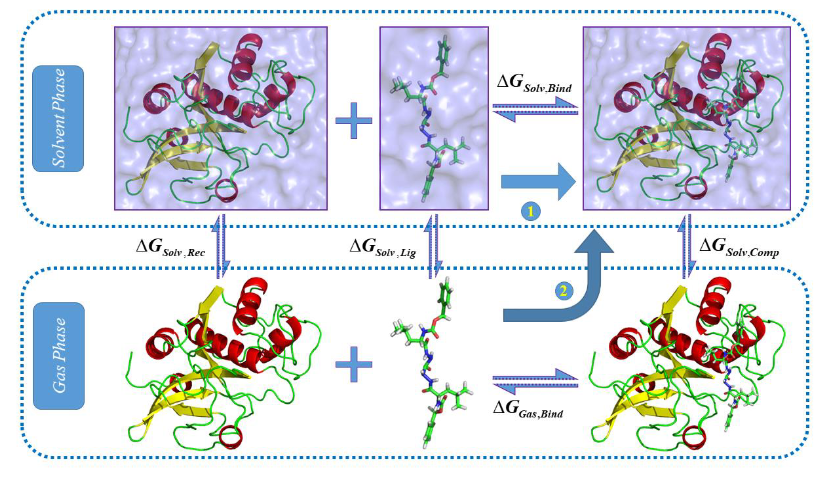
MM/PB(GB)SA 计算结合自由能的思想如图所示[2]。路径 1 描述的是实际中受体和配体中结合的方式:两者处于液态环境中并结合成复合物,其中 ΔGSolv, bind 为所求的结合自由能。考虑到在液相中两者结合的过程异常复杂,MM/PB(GB)SA 通过路径 2 简化结合过程:配体与受体先在气相中结合,然后将结合后的复合物置于液体中。从图中可以看出。两者的路径始末状态相同。通过该热力学循环可以巧妙地避开液态环境中复杂的能量变化,使得生物大分子体系中的结合自由能的计算变得可行且简便。其中热力学公式如下:
$$
\Delta G_{solv,bind}=\Delta G_{Gas,Comp}-\Delta G_{Gas,Rec}-\Delta G_{Gas,Lig}+\Delta G_{Solv,Comp}-\Delta G_{Solv,Rec}-\Delta G_{Solv,Lig}\
$$
$$
\Delta G_{bind}=\Delta H-TdS\
$$
$$
\Delta H=\Delta E_{MM}+\Delta G_{sol} \
$$
$$
\Delta G_{sol}=\Delta G_{pol}+\Delta G_{nonpol}\
$$
$$
\Delta E_{MM}=\Delta E_{elec}+\Delta {vdw}+\Delta E{int} \
$$
$$
\Delta G_{bind}=\Delta E_{elec}+\Delta {vdw}+\Delta G{pol}+\Delta G_{nonpol}-TdS
$$
焓变($\Delta H$)由气相能($\Delta E_{MM}$)和溶剂化自由能($\Delta G_{sol}$)组成
气相能
气相能($\Delta E_{MM}$)由静电项($\Delta E_{elec}$)、范德华项($\Delta E_{vdw}$)和内能项($\Delta E_{int}$)组成,内能项由键长、键角、二面角贡献,因为计算时蛋白-配体的结构是刚性不变的,因此计算过程中相互抵消,即$\Delta E_{int}=0$。因此气相能只需计算静电和范德华作用,计算公式如下:
$$
\Delta E_{elec}=\Delta E_{elec}^{Comp}-\Delta E_{elec}^{Pro}-\Delta E_{elec}^{Lig} \
$$
$$
\Delta E_{vdw}=\Delta E_{vdw}^{Comp}-\Delta E_{vdw}^{Pro}-\Delta E_{vdw}^{Lig} \
$$
其分别代表复合物、蛋白、配体的静电或者范德华能。
对于静电相互作用可以使用如下公式计算,其中$f=\frac{1}{4\pi \epsilon 0}=138.935458$(见gmx手册5.5),$q_i$为原子所带电荷,$r{ij}$为原子间距离,$\epsilon r$为溶质介电常数,$\epsilon 0$为真空介电常数:
$$
E{elec}=f\frac{q_i q_j}{\epsilon {r}r{ij}}
$$
在实际计算中,因为组分带有净电荷,气相的静电相互作用$E{elec}$往往会很大, 从而导致总的结合能绝对数值过大或为正值,可以使用德拜-休克尔屏蔽方法计算$E_{elec}$: 计算$E_{elec}$时使用考虑离子强度, 使用德拜特征长度对静电作用进行指数衰减. 这样处理后, 所得$E_{elec}$贡献就变小了[3]。
$$
E_{elec}=f\frac{q_i q_j}{\epsilon {r}r{ij}}e^{-\frac{r_{ij}}{\lambda D}}\
$$
$$
\lambda D =\sqrt{\frac{\epsilon{0}\epsilon{s}k_{B}T}{\sum {i} c_i e^2 z^{2}{i}}} \
$$
$$
\epsilon _s: 溶剂介电常数 \qquad c_i / z_i:离子浓度与电荷 \qquad e:电荷量 \qquad k_B: 玻尔兹曼常数
$$
对于范德华相互作用可以使用如下公式计算,其中$C_{ij}^{6}、C_{ij}^{12}$ LJ参数可以从生成的tpr文件中直接读取,不需要考虑top设定的结合规则进行转换,:
$$
E_{vdw}=\frac{C_{ij}^{(12)}}{r_{ij}^{12}}-\frac{C_{ij}^{(6)}}{r_{ij}^{6}} \
$$
$$
C_{ij}^{(6)}=4\epsilon _{ij} \sigma _{ij} ^6 \
$$
$$
C_{ij}^{(12)}=4\epsilon _{ij} \sigma _{ij} ^{12} \
$$
$$
\epsilon {ij} =\frac{1}{4}C{ij}^{(6)}\sigma _{ij} ^{-6} \
$$
$$
\sigma {ij}= \left(\frac{C{ij}^{(12)}}{C_{ij}^{(6)}}\right)^{-6}
$$
溶剂化自由能
$$
\Delta G_{sol}=\Delta G_{pol}+\Delta G_{nonpol}
$$
溶剂化自由能由极性溶剂化自由能($\Delta G_{pol}$)和非极性溶剂化自由能($\Delta G_{nonpol}$)组成。对于极性溶剂化自由能($\Delta G_{pol}$)可以使用广义波恩模型求解,也可以通过求解泊松玻尔兹曼方程(Poisson-Boltzmann )获得,在此处使用apbs求解PB方程。
对于apbs求解PB方程,需要考虑原子的半径,溶质溶剂的介电常数,格点的设定,网格长度,粗细格点中心,格点长度等等。格点的选择与长度设定参考了李老师的脚本,在实际计算中可能会出现vc_vecpmg: trapped exp overflows的警告,经过查询发现与网格的选择有关,后续需要继续优化。
原子半径对于求解也会有部分影响,可以根据$\left(\frac{C_{ij}^{6}}{C_{ij}^{12}}\right)^{\frac{1}{6}}$计算半径,也可以使用mbondi、mbondi2等其他原子半径。此处使用mbondi。
溶质溶剂介电常数分别选为2和78.4,溶质介电常数一般取值2-4。溶质介电常数的选取对于某些体系的计算影响甚大。高度带电的结合位点需要使用更高的介电常数。更进一步的,介电常数取值可以根据氨基酸不同而不同。
对于非极性溶剂化自由能$\Delta G_{nonpol}$通过溶剂可及表面积(SASA)计算,$\gamma$和$\beta$为经验常数,根据不同的模型,有不同的选择,此处选择值为:0.02267788和3.84928。
$$
\Delta G_{nonpol}=\gamma\cdot SASA + \beta
$$
相互作用熵
相互作用熵可以通过Normal mode分析获得,但是Normal mode过于耗时,此处选用张增辉组开发的相互作用熵方法进行计算[4],该方法通过对MM能量的取平均获得相互作用熵,计算的效率与精度均有大幅提高。
$$
-T \Delta S=KT ln\left<e^{\beta\Delta E_{pl}^{int}}\right> \
$$
$$
\left<E_{pl}^{int}=\frac{1}{T} \int_{0}^{T}E_{pl}^{int}(t)dt=\frac{1}{N}\sum_{i=1}^{N}E_{pl}^{int}(t_i)\right> \
$$
$$
\left<e^{\beta \Delta E_{pl}^{int}}\right>=\frac{1}{N}\sum_{i=1}^{N}e^{\beta\Delta E_{pl}^{int}(t_{i})} \
$$
$$
-T\Delta S=KTln\frac{1}{N}\sum_{i=1}^{N}e^{\beta\Delta E_{pl}^{int}(t_{i})}
$$
$E_{pl}^{int}$相互作用能包含蛋白配体之间的静电相互作用和范德华作用,$\left<E_{pl}^{int}\right>$为平均值,K为摩尔气体常数,$\beta=\frac{1}{KT}$
APBS并行
APBS开发文档写了可以并行,但是需要动态编译,具体的操作细节可以见我师兄写的APBS v3.4.1 的并行版编译[5] 。同时该并行指的是对于单帧结构计算,当机器拥有较多的核心时,使用多个核跑一帧会造成资源浪费和效率的降低。因此需要合理分配核心计算多帧结构,此处使用python的multiprocessing库进行多核并行。值得注意的是,APBS的系统调度有问题,即使将每个任务都进行了内核绑定,同时计算的效率也无法达到预期效果,但是速度也比单核跑快了一倍以上。
结果输出
因为懒,所以用matplotlib将结果可视化输出了。结果图片由多个子图组成的一张大图和单独保存的子图构成。所有的结合能数据以及残基分解数据都被保存到了csv文件中,命令行也会输出结合能的总结,详情见下节。
使用方法
python脚本及例子见我的github仓库[6]
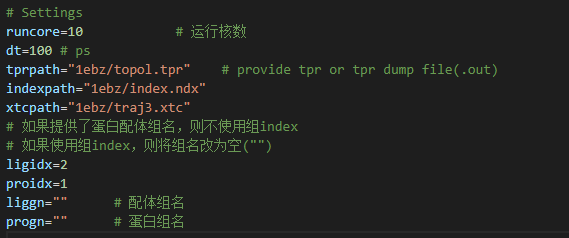
下载文件之后,打开MMPBSAMultiProcessV1.7.py,修改运行核数runcore、帧时间间隔dt、tpr路径tprpath、index路径indexpath、xtc路径xtcpath以及蛋白配体的index号。ligidx,proidx,见下图。值得注意的是,如果提供了蛋白配体组名,就默认不使用index,如果使用了组index,就必须将组名改为空"",可以根据自己的需要修改开头的apbs计算参数以及绘图的细节。
修改好参数,运行如下命令即可:
1 | python MMPBSAMultiProcessV1.7.py |
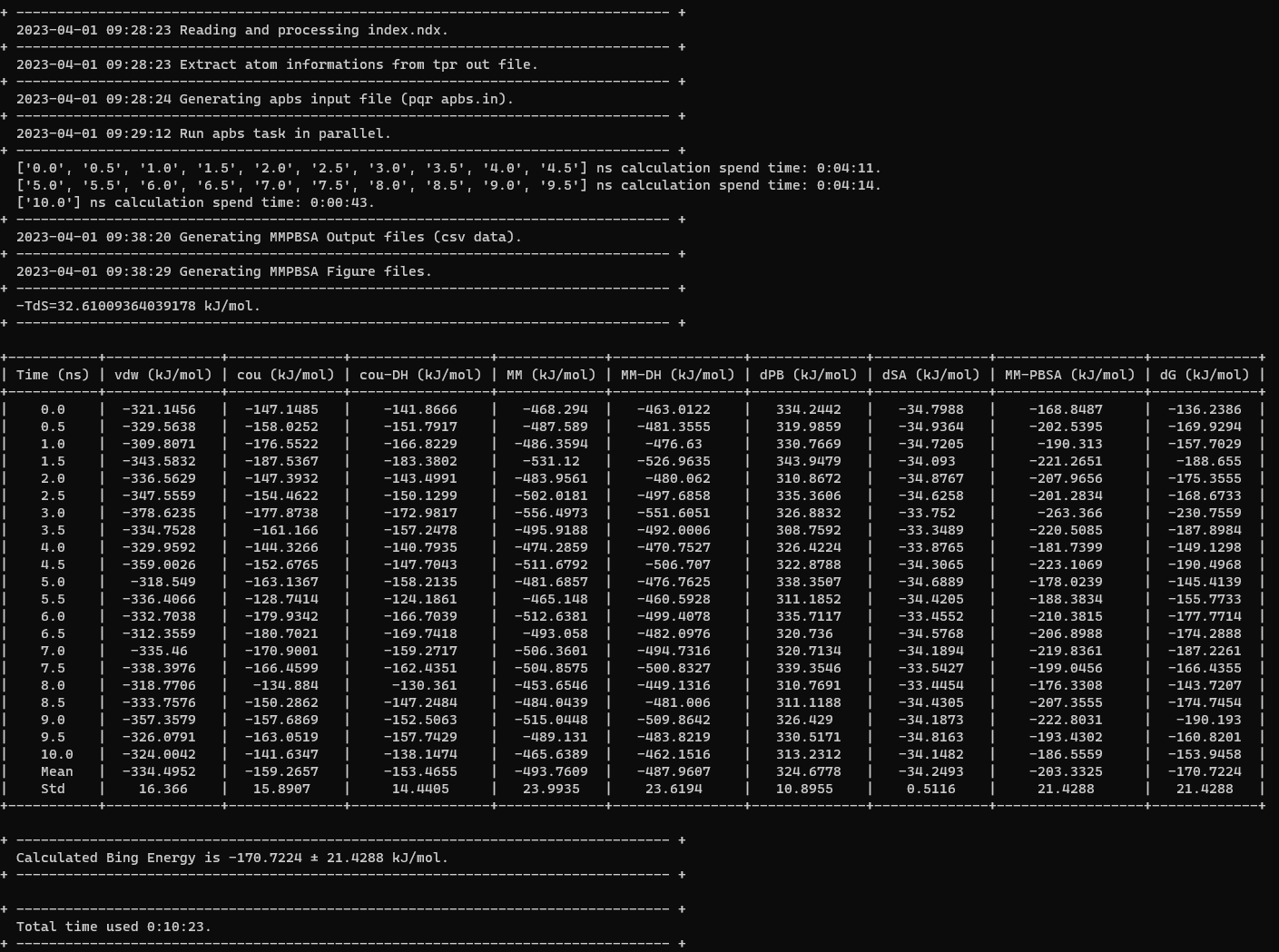
当所需库均安装完成,命令行可获得每步耗时、计算的构象熵变、结合能、总耗时等等。
工作目录内会生成残基不同能量项的平均值表avgresMMPBSA.csv,不同时间的残基能量项表TotalresDecomps.csv以及MMPBSA的汇总数据表TotalMMPBSA.csv。
工作目录也会根据数据生成不同的数据图。
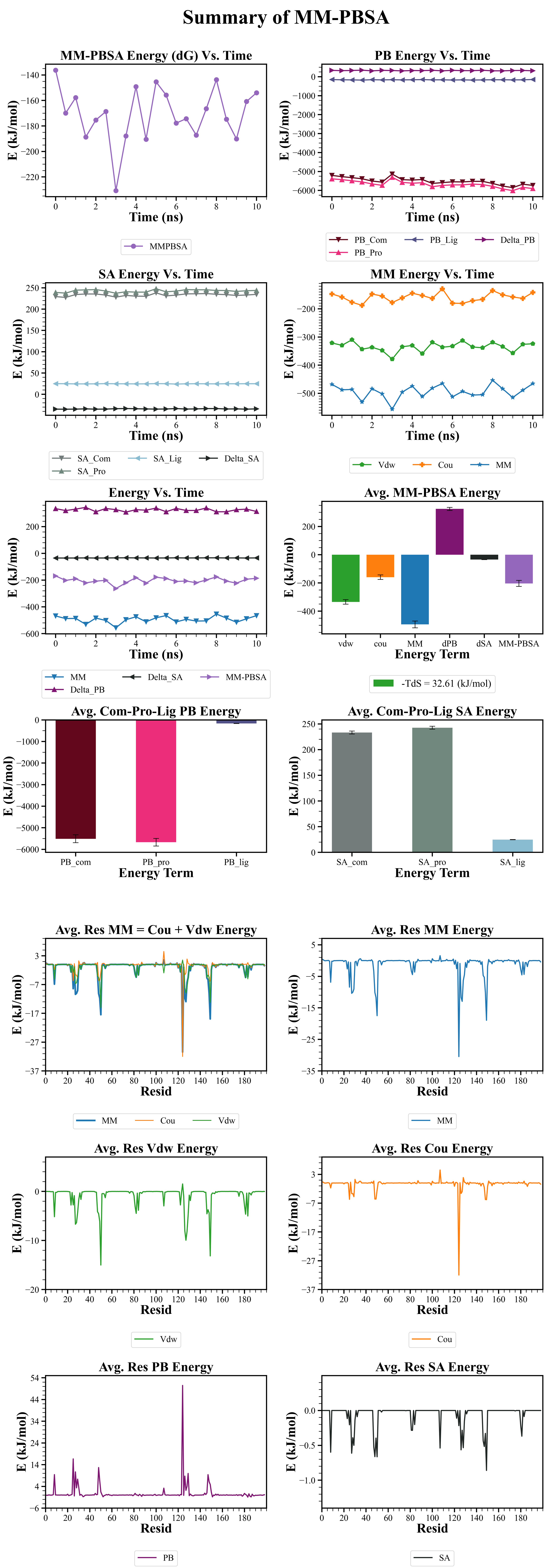
小结
本文算是个人学习mmpbsa的一个小小总结。整个工作的workflow如下:1)使用gmx dump将tpr文件转为可读文件,读取转换文件获得原子、残基的信息;2)使用mdanalysis读取轨迹,结合index计算MM能量,并生成蛋白、配体、复合物的apbs计算文件;3)并行调用apbs计算生成结果;4)提取apbs计算结果并合并总结;5)生成图片、表格并输出相关信息。
目前来看,计算的结果与李老师的脚本进行对比,相差不大。因个人水平有限,该脚本目前存在着如下一些问题:
- 格点处理划分存在误差,会出现警告
- 当输入未经处理的轨迹时,因内部算法的问题,会出现生成计算距离的数组需要内存量过大的报错。目前的解决方法是强制只生成包含蛋白和配体的数据(一般情况不会报错,如果遇到蛋白配体的编号不连续可能会出错)
- 不能指定开始与结束帧
- apbs并行速度需要优化
- 。。。
科研本就图一乐,但道阻且长!
Todo
- 自由指定计算的时间
- 将计算结果写入到pdb文件(类似于B因子)
- 支持GB模型
- 轨迹pbc处理
- 支持不同的半径类型
- 支持使用文件指定参数
- 绘制表面静电势并出图
- 算法、速度、步骤优化
- 。。。
参考链接
[1] https://jerkwin.github.io/gmxtools/
[2] 基于 MM/PBSA 和相互作用熵理论优化 结合自由能计算及其应用 黄开放
[3] https://jerkwin.github.io/2021/03/16/gmx_mmpbsa脚本更新-屏蔽效应与熵贡献
[4] Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein−Ligand Binding Free Energy
[5] https://zhuanlan.zhihu.com/p/618313803
[6] https://github.com/Zuttergutao/GMXAnalysis/tree/main/MMPBSAv1.7